Introduction
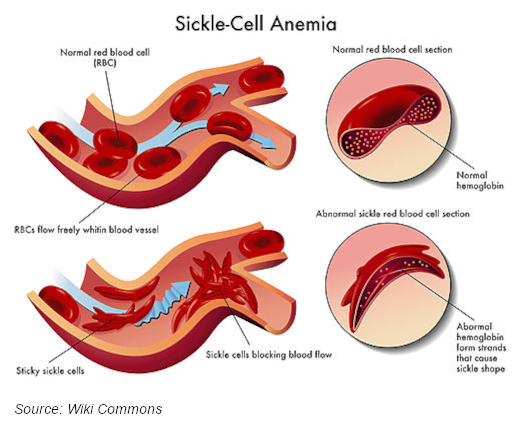
Sickle cell anemia is the most common inherited blood disorder in the United States affecting an estimated 100,000 people. The disease can cause a multitude of symptoms including anemia, episodes of pain, frequent infections, swelling of hands and feet, vision problems, and delayed growth/puberty. These symptoms range from mild to severe enough that the person has to be hospitalized. The disease affects mostly people who have ancestors from Africa, the Mediterranean, and South America. For many people, this disease can be extremely debilitating. This post will discuss hemoglobin and its function, the cause of sickle cell anemia, and testing and detection.
Hemoglobin and Its Function
Before discussing sickle cell disease itself, it would be helpful to understand the basic structure and function of a normal adult hemoglobin protein. The main purpose of hemoglobin is to transport oxygen from the lungs to all areas of the body and to transport CO2 from the tissues and extremities back to the lungs to be expelled. It is synthesized in the erythrocyte-producing cells of the bone marrow and is the main component of a person’s red blood cells. Normal adult hemoglobin (called Hemoglobin A or HbA) is composed of 4 polypeptide chains; two alpha chains and two beta chains.
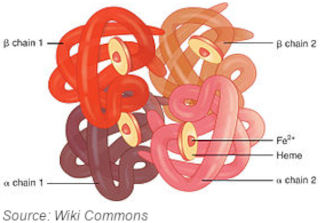
In the center of these chains is a heme molecule composed of a molecule of protoporphyrin IX and a ferrous ion. It’s the heme portion that actually binds the oxygen. The 4 polypeptide chains in a hemoglobin molecule each have a heme molecule associated with them. Therefore, one hemoglobin protein can bind four molecules of oxygen and four molecules of CO2 (in the form of bicarbonate ions). When the hemoglobin polypeptide chains become deformed, this can severely limit the body’s ability to do this. Generally, the polypeptide chains become deformed at the end of a red blood cell’s life and are destroyed/recycled in the liver and spleen.
Description
Cause
Sickle cell disease is is inherited and caused by a mutation in the HBB gene that codes for the beta globulin chain in the hemoglobin molecule. This mutation generates a single amino acid substitution in the sixth position of the beta globulin chain (a valine replaces a glutamine), causing the hemoglobin to be deformed and the red blood cells to curve or sickle. This mutated hemoglobin is known as Hemoglobin S (Hb S). The deformed cells can cause problems for the afflicted individual, blocking blood vessels and causing severe complications.
Complications and Early Signs & Symptoms
The signs and symptoms of sickle cell disease vary from person to person and usually become apparent around five to six months of age. These include jaundice, swelling of hands and feet, and fatigue. Because these symptoms can be indicative of other diseases, it is helpful that the doctor knows the full medical history of both parents.
As individuals with sickle cell disease age they tend to develop other complications, some being due to the disease itself and some as the side effects of the treatment. The main complication of this disease is anemia (hence the other name sickle cell anemia). This is due to the fact that the spleen sees these cells as damaged and tries to remove them. Removing this many red blood cells can cause the spleen to become severely enlarged. Not only does this cause the person severe discomfort, it can also cause them to become more susceptible to infections due to the important function the spleen serves in the immune system.
Due to the inappropriate shape of the red blood cells, other blood vessels can become blocked, causing the individual to experience bouts of extreme pain, vision problems, heart problems, leg ulcers, and stroke.
Difference Between Sickle Cell Disease and Trait
In order for a person to have full-blown sickle cell disease, they have to inherit a Hemoglobin S gene from both parents (S/S). This means that the person will have a more severe form of the disease and more complications.
If a person inherits a Hemoglobin S gene from one parent and a Hemoglobin A gene from the other parent, then they have what is called sickle cell trait (A/S). Sickle cell trait is typically milder than sickle cell disease, with little to no symptoms.
Testing/Detection
This section will discuss the three types of tests for sickle cell disease/trait that are most commonly used. There is a screening test, a confirmatory test and a genetic test.
Screening Test
The screening test for sickle cell disease/trait is fairly simple. It is a qualitative test based on the insolubility of Hemoglobin S compared to Hemoglobin A. In this test, if the hemoglobin is insoluble, a cloudy, turbid suspension is formed, which means that a hemoglobinopothy is present. A positive test may indicate the sickle cell disease (Hemoglobin S/S), sickle cell trait (Hemoglobin S/A), or Hemoglobin S combined with another hemoglobin variant. This test needs to be confirmed with other laboratory tests to verify the actual type of hemoglobin that is present, as conditions other than sickle cell can cause a positive result (multiple myeloma, polycythemia, or cryoglobulinemia).
HPLC
HPLC stands for high performance liquid chromatography. This is the test that is used to confirm the presence of hemoglobin S. In HPLC, compounds are separated based on mass. The analyte is distributed between a mobile phase and a stationary phase. As the analyte moves through the stationary phase, the molecules become separated according to mass. The stationary phase is generally a column consisting of some sort of packing material. As the analyte moves through the column, the larger molecules travel a shorter distance than smaller molecules. Once the test is completed, the position of the molecules determines the identity of the analyte. This is how Hgb S is distinguished from Hgb A by the position of the hemoglobin molecules in the column.
Genetic Test
Sickle cell disease or trait can be detected via genetic analysis. If a physician suspects that there is a possibility a baby may be born with sickle cell disease, an amniocentesis can be performed while the fetus is in utero or shortly after birth. This particular test detects the Hemoglobin S gene rather than the actual deformed hemoglobin. This enables the physician and family of the child to know right away if the child has sickle cell disease, as many of the symptoms can be present in other diseases and hemoglobinopathies.
Conclusion
As one of the most common hemoglobinopathies in the world, sickle cell disease is well understood. It is easy to diagnose due to the fact that it is so common. Sickle cell disease at this time does not have any treatment. Any treatment that the patient receives presently pertains to the management of symptoms. Unfortunately, even with the treatments available today, the life expectancy of someone with sickle cell disease is only between 42-47 years. As with any disease, proper management and regular doctor visits can help outcomes.